Authors:
(1) JunJie Wee, Department of Mathematics, Michigan State University;
(2) Jiahui Chen, Department of Mathematical Sciences, University of Arkansas;
(3) Kelin Xia, Division of Mathematical Sciences, School of Physical and Mathematical Sciences Nanyang Technological University & [email protected];
(4)Guo-Wei Wei, 1Department of Mathematics, Michigan State University, Department of Biochemistry and Molecular Biology, Michigan State University, Department of Electrical and Computer Engineering, Michigan State University & [email protected].
Table of Links
Abstract & Introduction
Results
Discussion
Conclusion
Materials and Methods
Software and resources, Code and Data Availability
Supporting Information, Acknowledgments & References
Abstract
Protein mutations can significantly influence protein solubility, which results in altered protein functions and leads to various diseases. Despite of tremendous effort, machine learning prediction of protein solubility changes upon mutation remains a challenging task as indicated by the poor scores of normalized Correct Prediction Ratio (CPR). Part of the challenge stems from the fact that there is no three-dimensional (3D) structures for the wild-type and mutant proteins. This work integrates persistent Laplacians and pre-trained Transformer for the task. The Transformer, pretrained with hunderds of millions of protein sequences, embeds wild-type and mutant sequences, while persistent Laplacians track the topological invariant change and homotopic shape evolution induced by mutations in 3D protein structures, which are rendered from AlphaFold2. The resulting machine learning model was trained on an extensive data set labeled with three solubility types. Our model outperforms all existing predictive methods and improves the state-of-the-art up to 15%
Keywords: Mutation, Protein solubility, Persistent Laplacian, Transformer, AlphaFold2.
1 Introduction
Genetic mutations alter the genome sequence, leading to changes in the corresponding amino acid sequence of a protein. These alternations have far-reaching implications on the protein’s structure, function, and stability, affecting attributes such as folding stability, binding affinity, and solubility. The consequences of protein mutations have been extensively studied in diverse fields such as evolutionary biology, cancer biology, immunology, directed evolution, and protein engineering [1]. Understanding the impact of genetic mutations on protein solubility is crucial in various fields, including protein engineering, drug discovery, and biotechnology. Accurately analyzing and predicting the impact of mutations on protein solubility is therefore crucial in many fields, facilitating the engineering of proteins with desirable functions. There are numerous intricately interconnected factors impacting protein solubility, ranging from amino acid sequence arrangement, post-translational modifications, protein-protein interactions, to environmental conditions, such as solvent type, ion type and concentration, the presence of small molecules, temperature, etc. Unfortunately, the existing data set does not contain sufficient information. This complexity poses significant challenges for the accurate prediction and modeling of protein solubility, often requiring multifaceted computational approaches for reliable outcomes.
Computational predictions serve as a valuable complement to experimental mutagenesis analysis of protein stability changes upon mutation. Such computational approaches offer several advantages, including being economical, efficient, and provide a viable alternative to laborintensive site-directed mutagenesis experiments [2]. As a result, the development of accurate and reliable computational techniques for mutational impact prediction could substantially enhance the throughput and accessibility of research in protein engineering and drug discovery
Over the years, a variety of computational methods have been developed to explore the effects of mutations on protein solubility, including but not limited to CamSol [3], OptSolMut [4], PON-Sol [5], SODA [6], Solubis [7], and others as summarized in a recent review [8]. CamSol employs an algorithm to construct a residue-specific solubility profile, although no explicit method has been made publicly available. OptSolMut is trained on 137 samples, each featuring single or multiple mutations affecting solubility or aggregation. PON-Sol utilizes a random forest model trained on a dataset of 406 single amino acid substitutions labeled as solubilityincreasing, solubility-decreasing, or exhibiting no change in solubility. SODA, which is based on the PON-Sol data, specifically targets samples with decreasing solubility [6]. Solubis is an optimization tool that increases protein solubility and integrates interaction analysis from FoldX [2], aggregation prediction from TANGO [9], and structural analysis from YASARA [10]. Recently, PON-Sol2 [11] extended the original PON-Sol dataset and employed a gradient boosting algorithm for sequence-based predictions. Despite of intensive effort, the current prediction accuracy in terms of normalized Correct Prediction Ratio (CPR) remains very low, calling for innovative strategies.
Topological data analysis (TDA) is a relatively new approach for data science. Its main technique is persistent homology [12, 13]. The essential idea of persistent homology is to construct a multiscale analysis of data in terms of topological invariants. The resulting changes of topological invariants over scales can be used to characterize the intricate structures of data, leading to an unusually powerful approach in describing protein structure, flexibility, and folding [14]. Persistent homology was integrated with machine learning for the classification of proteins in 2015 [15], which was one the first integrations of TDA and machine learning, and the predictions of mutation-induced protein stability changes [16, 17] and protein-protein binding free energy changes [18, 19]. One of the major achievements of TDA is its winning of D3R Grand Challenges, an annual worldwide competition series in computer-aided drug design [20, 21]. A nearly comprehensive summary of the early success of TDA in biological science was given in a review [22].
However, persistent homology only tracks the changes in topological invariants and cannot capture homotopic shape evolution of data over scales or induced by mutations. To overcome this limitation, Wei and coworkers introduced persistent combinatorial Laplacians, also called persistent spectral graphs, on point clouds [23] and evolutionary de Rham-Hodge method on manifolds [24] in 2019. The essence of these methods is the persistent topological Laplacians (PTLs) either on point clouds or on manifolds. PTLs not only fully capture the topological invariants in its harmonic spectra as those given by persistent homology, but also capture the homotopic shape evolution of data during the multiscale analysis or a mutation process. PTLs were applied to the predictions of protein flexibility [25] and protein-ligand binding free energies [26], protein–protein interactions[27, 28], and protein engineering [1]. The most remarkable accomplishment by persistent Laplacian is its accurate forecasting of emerging dominant SARS-CoV-2 variants BA.4 and BA.5 about two months in advance [29].
However, TDA approaches depend on the biomolcular structures, which may not be available. In fact, many proteins involved in the present study do not have 3D structures. In recent years, advanced natural language processing (NLP) models, including Transformers and long short-term memory (LSTM), have been widely implemented across various domains to extract protein sequence information. For example, Tasks Assessing Protein Embedding (TAPE) introduced three different architectures, namely transformer, dilated residual network (ResNet), and LSTM [30]. Additionally, LSTM-based models like Bepler [31] and UniRep [32] have been developed. Additionally, large-scale protein transformer models trained on extensive datasets comprising hundreds of millions of sequences have made significant advancements in the field. These models, including Evolutionary Scale Modeling (ESM) [33] and ProtTrans [34, 35], have exhibited exceptional performance in capturing a variety of protein properties. ESM, for instance, allows for fine-tuning based on either downstream task data or local multiple sequence alignments [36]. In the present work, we leverage the pre-trained ESM transformer model to extract crucial information from protein sequences.
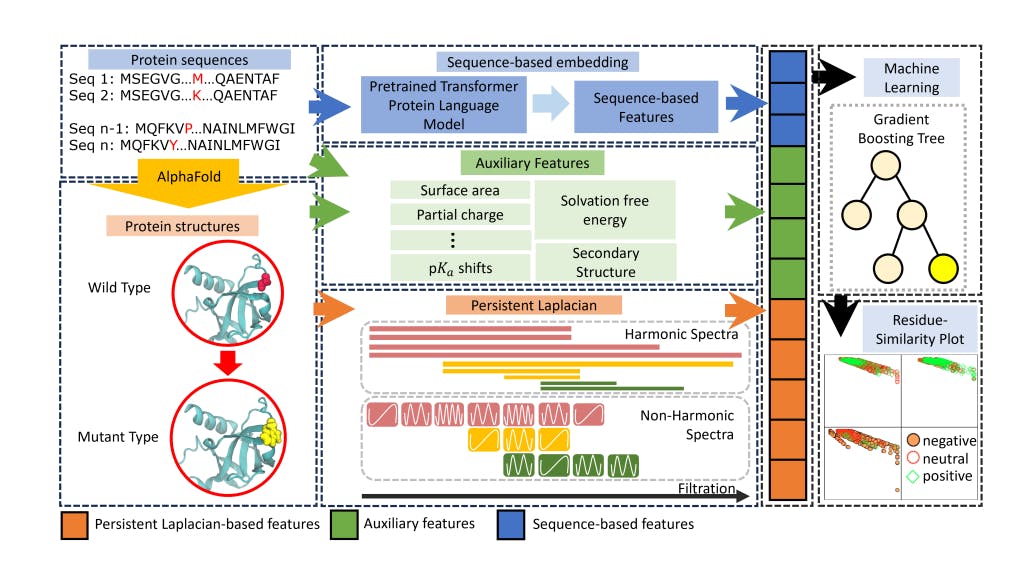
In this work, we will integrate transformer-based sequence embedding and persistent topological Laplacians to predict protein solubility changes upon mutation. While sequence-based models can be applied without 3D structural information, the PTL-based features require highquality structures. We generate 3D structures of wild type proteins from AlphaFold2 [37] to facilitate topological embedding. By combining both embedding approaches, they naturally complement each other in classifying protein solubility changes upon mutation. These embeddings are fed into an ensemble classifier, gradient boosted trees (GBT), to build a machine learning model, called TopLapGBT. We validate TopLapGBT on the classification of protein solubility changes upon mutation. We demonstrate that this integrated machine learning model gives rise to a substantial improvement as compared to existing state-of-the-art models. Residue-Similarity plots are also applied to assess how well the TopLapGBT model classify three solubility labels.
This paper is under CC 4.0 license.